Determinar la secuencia del genoma de células bacterianas individuales siempre ha sido un desafío técnico debido a un sesgo aparentemente inmutable en la etapa de amplificación de genes del proceso, lo que dificulta producir una secuencia de genoma de alta cobertura a partir de una sola célula bacteriana.
Un nuevo proceso desarrollado por investigadores del Centro de Células Únicas, Instituto de Bioenergía y Tecnología de Bioprocesos de Qingdao (QIBEBT) de la Academia de Ciencias de China (CAS), reduce significativamente este sesgo, abriendo nuevos horizontes para la investigación de células individuales. Su estudio ha sido publicado en Fronteras en bioingeniería y biotecnología El 29 de junio.
Cuando se utilizan métodos tradicionales de secuenciación genética, es normal utilizar millones de células al mismo tiempo. Pero tal secuenciación masiva de células inevitablemente borra las diferencias entre las células, lo que dificulta la exploración de las relaciones entre diferentes linajes celulares o la realización de cualquier otro tipo de investigación que se base en el estudio de una célula a la vez. Estos desafíos tecnológicos son particularmente peligrosos para las bacterias, porque la célula bacteriana es tan pequeña que su contenido de ADN es varias veces menor que el de una célula humana o animal.
Para secuenciar un gen o un genoma completo, primero se debe «amplificar» el ADN. La amplificación de genes, a veces llamada «fototranscripción molecular», puede producir millones o incluso miles de millones de copias de fragmentos de ADN.
La amplificación plantea pocos problemas para la secuenciación de genes a granel. Sin embargo, en la amplificación de ADN de una sola célula bacteriana, especialmente en genomas completos, la cantidad de ADN disponible para la transcripción es muy limitada y el proceso de amplificación puede introducir sesgos que sobrerrepresentan o subrepresentan diferentes regiones del genoma.
«Reducir el sesgo de amplificación ha sido algo sagrado para los investigadores de la amplificación del genoma unicelular», dijo Zhang Jia, biotecnólogo de QIBEBT y autor principal del artículo. Creemos que ahora hemos encontrado una manera de abordar este problema.
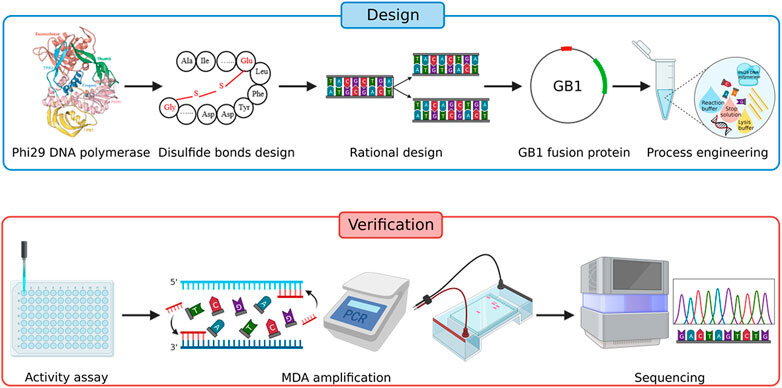
Los investigadores desarrollaron un proceso llamado amplificación mejorada del genoma unicelular (iSGA). Implica principalmente la ingeniería de proteínas, así como la ingeniería de procesos para la capacidad de amplificación de la ADN polimerasa phi29. La ADN polimerasa phi29 se usa comúnmente para la amplificación del genoma completo de una sola célula, pero adolece de problemas de baja cobertura del genoma y baja eficiencia.
«La ADN polimerasa iSGA phi29 que desarrollamos es más del doble de eficiente y robusta que la ADN polimerasa phi29 convencional», dijo Zhang Jia. «También es unas 11 veces más barata que la versión comercial convencional comúnmente disponible».
Los investigadores lograron esto modificando la polimerasa para mejorar su capacidad de amplificación. Usando un método llamado autorreplicación fragmentada (CSR) para desarrollar la ADN polimerasa phi29, modificaron su estructura para mejorar su actividad, especificidad y estabilidad. «Nuestra ADN polimerasa recientemente desarrollada, a la que llamamos ADN polimerasa HotJa Phi29, muestra una cobertura genómica significativamente mejor (99,75 %) a una temperatura más alta (40 ℃) en comparación con las polimerasas comerciales existentes», dijo el autor principal, el profesor Xu Jian, del Centro de Células Únicas de QIBEBT.
También optimizaron las condiciones para la reacción de amplificación y modificaron el tampón de amplificación (la solución que proporciona un entorno químico adecuado para la ADN polimerasa) para mejorar la estabilidad y la actividad de la ADN polimerasa phi29. Además, los investigadores desarrollaron un método de descontaminación más eficiente para reducir la contaminación durante el proceso de amplificación.
Los investigadores ahora planean mejorar el método de polimerasa de ADN iSGA y HotJa Phi29 en varias aplicaciones y tipos de muestras, reduciendo aún más los costos. El equipo ha desarrollado una serie de herramientas que incluyen RACS-Seq, FlowRACS y EasySort para la clasificación funcional de células microbianas individuales. Su objetivo, al desarrollar estas herramientas y reactivos, es permitir que los microbiólogos clasifiquen y secuencien cualquier célula microbiana individual en la Tierra, sin preocuparse por la calidad de los datos o el costo de los reactivos.
más información:
Jia Zhang et al, Amplificación mejorada del genoma unicelular mediante polimerasa de ADN phi29 de alta eficiencia, Fronteras en bioingeniería y biotecnología (2023). DOI: 10.3389/fbioe.2023.1233856

«Solucionador de problemas. Gurú de los zombis. Entusiasta de Internet. Defensor de los viajes sin disculpas. Organizador. Lector. Aficionado al alcohol».